





R&D
Polyterpenes by ring opening metathesis polymerization of caryophyllene and humulene
R&D
Received 11th February 2013,
Accepted 15th March 2013
DOI: 10.1039/c3gc40300a
www.rsc.org/greenchem
Ring opening metathesis polymerization of the natural sesquiterpenes caryophyllene and humulene, optionally complemented by exhaustive post-polymerization hydrogenation, yields non-crosslinked linear polymers with unprecedented microstructures reflecting the specific scaffolds of the feedstocks and with low glass transition temperatures in the range from −15 to −50 °C.
Polymers with their myriad functions are indispensable to any modern technology. Today, their production is largely based on fossil fuels. In view of their limited range, other sources are desirable on the long term. Beyond such considerations, renewable resources are also attractive as they can contain unique chemical structures. Thus, carbohydrates,1 fatty acids2 or terpenes3 contain a wealth of different structural elements. Particularly, terpenes and terpenoids feature thousands of different scaffolds.4 However, except for pinene, limonene and myrcene, there are few examples of other terpenes5 used for polymerization. To translate the structural elements of a given renewable resource into useful material properties, appropriate synthetic methods are required.
Caryophyllene and humulene are among the most abundant and cheapest sesquiterpenes. They are found in many plants and fungi.6 For example, more than 105 tons of clove oil7 are produced annually by the clove tree Eugenia caryophyllata (Syzygium aromaticum). Clove oil contains 7–12% caryophyllene and 1–4% humulene. Another potential source is hop oil, with up to 25% caryophyllene and 45% humulene.8
Concerning the utilization of caryophyllene or humulene as a source of polymers, only ill-defined polymeric materials have been obtained from such sesquiterpenes as undesired sideproducts during episulfidation.9 The unsaturated cyclic nature of caryophyllene and humulene in principle makes them suitable for ring opening metathesis polymerization (ROMP).10–13 Thus, (−)-trans-caryophyllene (or β-caryophyllene) features a cyclobutane ring fused in a trans fashion to a ninemembered ring containing a 1,5-diene with the intra-ring double bond in Earrangement. Humulene, (1E,5E,8E)-1,4,4,8- tetramethyl-cycloundeca-1,5,8-triene, possesses an elevenmembered ring with all three double bonds in a trans arrangement. ROMPs of such motifs are unusual, and there are only a few reports of ROMP of nine- and eleven-membered rings, namely of the parent unsubstituted cyclononene or cycloundecene.14 There is also only scarce evidence of ROMP of 1-methyl-substituted cycloalkenes.15
We now report on the reactivity of caryophyllene and humulene in metathesis polymerization, and on the properties and the reactivity of the polymers obtained (Scheme 1).
Five catalyst precursors differing in the nature of the active species and the initiation reactivity were screened for their behaviour towards caryophyllene, namely [(PCy3)2Cl2RuvCHPh] (1), [(PCy3)(η-C–C3H4N2Mes2)Cl2RuvCHPh] (2), [(3-BrPyr)(η-C–C3H4N2Mes2)Cl2RuvCHPh] (3), [(PCy3)Cl2RuvCH(o-iOPr–Ph)] (4) and [(η-C–C3H4N2Mes2)Cl2RuvCH(o-iOPr–Ph)] (5). Reactions were run at 25 °C without any solvent over a prolonged period of a week in order to ensure maximum conversion (Table S1†). Polycaryophyllene can be produced in good yield with catalyst precursors 2, 3 and 5. With 1st generation metathesis catalysts (1 and 4), no polymer was obtained whatever the catalyst/monomer ratio.16 For 2nd generation catalysts (2 and 5), the yield of the polymerization drastically decreased with lower catalyst loading, probably due to a deactivation/decomposition of the catalyst. By contrast, with 3, complete conversion could be achieved even at a low catalyst load of 0.04 mol%.
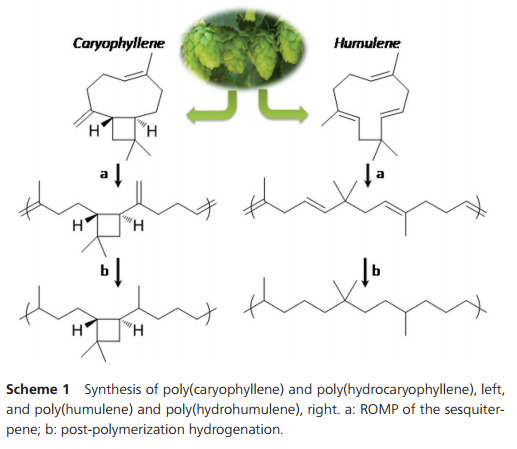
Scheme 1 Synthesis of poly(caryophyllene) and poly(hydrocaryophyllene), left, and poly(humulene) and poly(hydrohumulene), right. a: ROMP of the sesquiterpene; b: post-polymerization hydrogenation.
The progress of polymerizations over time with different catalyst precursors was also followed with 1 H NMR by monitoring the vCHR, vCH2 and CH3 resonances (Fig. 1a). Catalyst precursor 3 appears to be the most active for ROMP of caryophyllene, with total conversion reached in less than 12 hours. 2 initially polymerizes at a similar rate, but levels off after ca. 4 hours, indicating catalyst deactivation. Indeed, with 2nd generation catalysts (2 and 5), ruthenium black is formed during the polymerization. This was not observed with 3. ROMP with 5 has a very long induction time and the conversion starts only after 5 hours at a lower rate. The E/Z selectivity of metathesis appears to be dependent on the catalyst; with 2, 55% of E configured product was obtained vs. 63% with 5 and 87% with 3. However, this has no observable impact on the polymers Tg (vide infra).
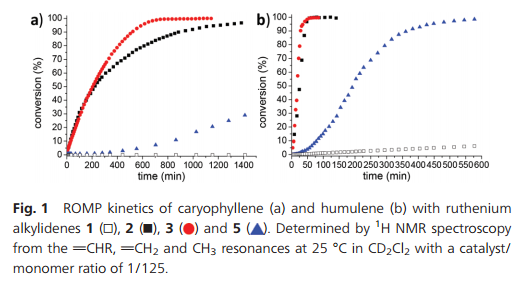
Fig. 1 ROMP kinetics of caryophyllene (a) and humulene (b) with ruthenium alkylidenes 1, 2, 3 and 5. Determined by 1H NMR spectroscopy from the vCHR, vCH2 and CH3 resonances at 25 °C in CD2Cl2 with a catalyst/monomer ratio of 1/125.
During polymerization, the exocyclic methylene group does not react to any observable extent. Thus, the ratio between =CHR and =CH2 protons remains constant during the polymerization. Also, the polymers formed are entirely soluble, which confirms that no cross-linking occurred. The polycaryophyllene microstructure depicted in Scheme 1 was confirmed by comprehensive 1H and 13C NMR analyses (Fig. 2 and ESI†). Only six olefinic 13C resonances are observed for the polycaryophyllene, confirming that neither head–head nor tail–tail propagation nor cross-linking occurs to an observable extent during the ROMP. The observed lack of reactivity of the vinylidene bond towards metathesis can be related to the high steric demand induced by the cyclobutane ring and the fact that the HOMO is mainly located on the other double bond. Finally, the intact cyclobutane ring present in the polymer is evidenced by its characteristic resonances at [(δ-1H) ppm/(δ-13C) ppm)] HC–CvCH2 [2.37/41.74], –CH2–CH [1.87/48.69], CH2 [1.77/39.83] and [1.45/39.83], and CMe2 [−/33.85]. The number average molecular weights (Mn) of the polycaryophyllene obtained with 3 were in the range of 1.7 to 2.0 × 104 g mol−1 (against polystyrene standards). Molecular weight distributions of Mw/Mn 1.8–1.9 point to a well-behaved nature of the polymerization reaction, and to chain transfer, presumably by back-biting, determining molecular weights. The latter is in line with the observed formation of several polymer chains per catalyst precursor molecule, as calculated from polymer molecular weights and catalyst loading. The polycaryophyllene obtained is a colorless, sticky and viscous material. DSC reveals a glass transition around Tg = −32 °C.
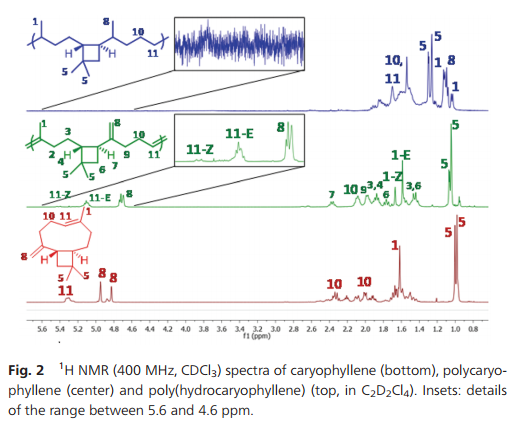
Fig. 2 1 H NMR (400 MHz, CDCl3) spectra of caryophyllene (bottom), polycaryophyllene (center) and poly(hydrocaryophyllene) (top, in C2D2Cl4). Insets: details of the range between 5.6 and 4.6 ppm.
The two different types of double bonds in polycaryophyllene may undergo undesired cleavage reactions, cross-linking or rearrangements upon storage or in applications. To this end, post-polymerization hydrogenation was studied. Exposure to 50 bar of H2 at 80 °C in the presence of a standard Pd/C catalyst (1/1 CH2Cl2–MeOH solvent) resulted in virtually complete conversion as evidenced by the absence of any olefinic 1H and 13C resonances (Fig. 2 and ESI†). This corresponds to a degree of hydrogenation >99.5%. In 1H NMR spectra four new doublets, corresponding to CH3–CH motifs resulting from the hydrogenation of the double bonds, appear at 1.10, 1.08, 1.06 and 1.00 ppm (respectively 19.90, 27.69, 17.28 and 16.56 ppm in 13C NMR spectra). For one of the new stereocenters resulting from the reduction of the CvCH2 motifs a high ratio of 1/10 of the two stereoisomers is observed, compared to a 1/2 ratio for the other newly formed stereocenter. This high stereoselectivity can be understood in that the chiral center of the cyclobutane ring significantly impacts the hydrogenation pathways of the adjacent vinylidene unit. Notably, the cyclobutane ring is not affected by the hydrogenation procedure.17 1H–13C HSQC demonstrated the presence of six additional CH carbon atoms, which agrees with the expected structure. Hydrogenation results in an increase of the Tg to ca. −16 °C.
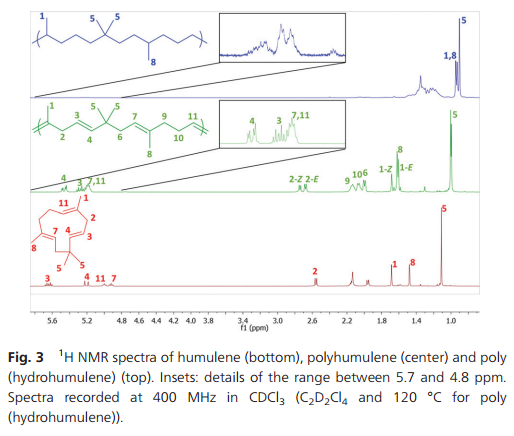
Fig. 3 1H NMR spectra of humulene (bottom), polyhumulene (center) and poly (hydrohumulene) (top). Insets: details of the range between 5.7 and 4.8 ppm. Spectra recorded at 400 MHz in CDCl3 (C2D2Cl4 and 120 °C for poly (hydrohumulene)).
Based on the aforementioned findings, polymerization of humulene was performed with 3 at 25 °C in bulk (Table S2†). Colorless sticky materials were obtained in high yield even at a low catalyst loading (0.04 mol%). Mn amounted to 3.0 × 104 g mol−1 typically, with Mw/Mn ≈ 2. DSC showed a Tg around −48 °C.
Microstructure analysis of the polymer indicates that the ROMP occurs only on one of the three different double bonds of the humulene starting material (Scheme 1 and Fig. 3). The two other double bonds strictly retain their E configuration in the polymer, while a 60/40 E/Z ratio is observed for the reacted double bond. Moreover, only one new 13C NMR resonance arises for a methyl in α position to a Z double bond (23.37 ppm vs. 16.24, 16.18 and 15.95 ppm). The methylene group adjacent to two double bonds is also strongly affected by the E/Z configurational change ([(δ-1H) ppm/(δ-13C) ppm]):[2.74/35.48] for Z and [2.67/43.27] for E. This indicates that the trisubstituted double bond adjacent to this methylene unit undergoes ROMP. The unreactivity of the disubstituted olefin can be understood in that the two methyl substituents in β position generate a high steric constraint. However, the steric constraints at the two trisubstituted double bonds appear to be similar. DFT studies provide an insight into the origin of the different reactivities observed. The HOMO of humulene is located mainly on the reacting double bond and is high in energy (+23 kJ mol−1 compared to the occupied orbital located on the other trisubstituted double bond) due to two antibonding π–π interactions with the other double bonds (cf. ESI†). Monitoring of humulene ROMP over time shows that the polymerization is faster compared to caryophyllene polymerization under the same conditions (Fig. 1b). Complete conversion is achieved in one hour using 2 or 3 as a catalyst precursor, and in ten hours with 5.18 Notably, ROMP of humulene also occurs with 1. However, the reaction is slow and after 12 hours the conversion is 10%.
Hydrogenation of the polyhumulene under the same conditions as polycaryophyllene resulted in a degree of hydrogenation of 98.3% (Fig. 3). A new 1H doublet resonance arises from the CH3–CH motif resulting from the hydrogenation of the trisubstituted double bonds, with δ = 0.93 ppm (19.86, 19.82 and 19.77 ppm in 13C NMR).19 DSC of polyhydrohumulene reveals a Tg around −44 °C.
The novel polymers obtained are soft materials with a low Tg. This renders them attractive for film formation and coatings. An established environmentally friendly route to polymer coatings and films is their generation from aqueous polymer dispersions. To this end, preliminary studies showed that ROMP of caryophyllene as well as the hydrogenation step could also be performed in aqueous emulsion using mini- or microemulsion techniques,20 without any additional organic solvents. For example, a solution of 3 in 3 mL of caryophyllene and 0.4 mL of hexadecane hydrophobe was ultrasonicated with 30 mL of an aqueous SDS solution (13.3 g L−1 ). Polymerization afforded a dispersion of particles with an average size of typically ca. 200 nm and a polydispersity of 0.1–0.3. NMR spectroscopy confirmed that the polymer produced is similar to the one produced in bulk. Also, molecular weights are comparable (Mn = 2.0 × 104 g mol−1 and Mw/Mn = 1.9).
In conclusion, polymers can be obtained by ROMP of naturally occurring terpenes. Namely, the readily available sesquiterpenes caryophyllene and humulene can be polymerized with appropriate ruthenium alkylidenes. Their nine- and elevenmembered rings, respectively, are opened with selective reaction of only one of the different types of double bonds present. This results in non-crosslinked polymers with a well-defined microstructure. These polymers as well as their fully saturated analogues are soft materials characterized by a glass transition in the range from −15 to −50 °C. This renders them attractive for environmentally friendly hydrophobic films and coatings based on renewable resources. To this end, aqueous dispersions are accessible by ROMP in emulsion as demonstrated with caryophyllene.
Notes and references
1 A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411; A. Gandini, Macromolecules, 2008, 41, 9491.
2 M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788; U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854.
3 P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8.
4 J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788.
5 J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 51, 11805.
6 I. G. Collado, J. R. Hanson and A. J. Macías-Sánchez, Nat. Prod. Rep., 1998, 15, 187.
7 http://faostat3.fao.org/home/index.html
8 E. Breitmaier, Terpenes, Wiley-VCH, Weinheim, 2006.
9 T. Ashitani, A.-K. Borg-Karlson, K. Fujita and S. Nagahama, Nat. Prod. Res., 2008, 22, 495.
10 For attempted synthesis of caryophyllene by ring closing metathesis: M. S. Dowling and C. D. Vanderwal, J. Org. Chem., 2010, 75, 6908.
11 Ring opening reactions via rearrangements and cyclobutane fragmentation of caryophyllene: W. K. Giersch, A. F. Boschung, L. Snowden and K. Schulte-Elte, Helv. Chim. Acta, 1994, 77, 36.
12 Cleavage of the endocyclic double bond of caryophyllene: A. F. Barrero, J. Molina, J. E. Oltra, J. Altarejos, A. Barrangán, A. Lara and M. Segura, Tetrahedron, 1995, 51, 3813; S. F. R. Hinkley, N. B. Perry and R. T. Weavers, Tetrahedron, 2005, 61, 3671.
13 For attempted ring closing metathesis in the synthesis of humulene: A. D. Rodriguez, E. Gonzalez and C. Ramirez, Tetrahedron, 1998, 54, 11683; T. Hu and E. J. Corey, Org. Lett., 2002, 4, 2441.
14 S. Warwel and H. Kätker, Synthesis, 1987, 935; B. Lebedev and N. Smirnova, Macromol. Chem. Phys., 1994, 195, 35.
15 S. R. Wilson and D. E. Schalk, J. Org. Chem., 1976, 41, 3929; C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903; J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 1383.
16 1st generation metathesis catalysts are known for their poor efficiency toward the synthesis of trisubstituted olefins. See for example: A. K. Chatterjee and R. H. Grubbs, Org. Lett., 1999, 1, 1751.
17 Moreover, the hydrogenation of caryophyllene monomer under similar conditions afforded bicyclic hydrocaryophyllene with its intact four-membered ring rather than monocyclic hydrohumulene.
18 After ROMP some isomerization of the double bonds of the polymer was observed with 2 as a catalyst precursor.
19 No selectivity was observed and a racemic mixture of the four stereoisomers was obtained.
20 V. Monteil, P. Wehrmann and S. Mecking, J. Am. Chem. Soc., 2005, 127, 14568.